allgemeine Pathologie
erbliche Krebserkrankungen
männliche Genitaltumore
Prostatakarzinom
Hodenkarzinom
weibliche Genitaltumore
Ovarialkarzinom
Corpuskarzinom
Cervixkarzinom
Vaginalkarzinom
Vulvakarzinom
Urothelkarzinome
Blasenkarzinom
Hauttumore
Lunge & Mediastinum
Magen-Darm-Tumore
Mammakarzinom / Brustkrebs
- Lymphödem bei Brustkrebs
Plasmozytom
Krebszentrum Heidelberg
onmeda
Humangenetik
Erbliche Krebserkrankungen
Zu den häufigsten erblichen Krebserkrankungen gehören erblicher Brust- und Eierstockkrebs sowie erblicher Darmkrebs.
Erblicher Brust- und Eierstockkrebs
Jede 8. bis 10. Frau erkrankt im Laufe ihres Lebens an Brustkrebs (Mamma-Karzinom), wobei die meisten Erkrankungen im höheren Alter auftreten. Erbliche Faktoren spielen eine wesentliche Rolle bei der Entstehung von Brustkrebs. So haben 25 % (jede vierte) der an Brustkrebs Erkrankten Familienangehörige, die ebenfalls an Brustkrebs erkrankt sind. Dabei bedeutet das Auftreten von Brust- und Eierstockkrebs (Ovarialkarzinom) in der Familie, auch bei mehreren Familienangehörigen, nicht von vornherein, dass eine erbliche Brust- und Eierstockkrebserkrankung vorliegt. Klarheit kann hier nur in einem genetischen Beratungsgespräch erlangt werden. Oft tragen diese ausführlichen Gespräche auch zur Beruhigung der Ratsuchenden bei.
Mindestens 5 % der Brustkrebserkrankungen sind jedoch klar erblich bedingt. Sie beruhen auf einer Mutation in einem der sog. Brustkrebsgene, die von einer Generation an die nächste weitergegeben werden kann. In etwa der Hälfte der Fälle liegen Mutationen in den Genen BRCA1 und BRCA2 vor.
Eine genetische Beratung ist bei folgenden Konstellationen in einer Familie sinnvoll:
- Zwei an Brustkrebs Erkrankte, davon eine unter 50 Jahren
- Drei oder mehr an Brustkrebs Erkrankte, unabhängig vom Alter
- Eine unter 30 Jahren an Brustkrebs Erkrankte
- Eine unter 40 Jahren an beidseitigem Brustkrebs Erkrankte
- Mindestens eine an Brustkrebs und eine an Eierstockkrebs Erkrankte
- Zwei oder mehr an Eierstockkrebs Erkrankte
- Mann mit Brustkrebs
Erblicher Darmkrebs
Dickdarm- und Enddarmkrebs (kolorektale Karzinome) machen etwa ein Drittel aller Krebserkrankungen aus. An Darmkrebs erkranken besonders Menschen in höherem Lebensalter. Wenn in einer Familie jüngere Menschen an Darmkrebs erkranken, liegt der Verdacht auf eine erbliche Form dieser Erkrankung nahe. Man geht heutzutage davon aus, dass etwa 5-10 % der Darmkrebserkrankungen erblich sind. Dabei unterscheidet man verschiedene Krankheitsbilder:
- das erbliche kolorektale Karzinom ohne Polyposis (HNPCC)
- die familiäre adenomatöse Polyposis (FAP)
HNPCC
Das erbliche kolorektale Karzinom ohne Polyposis (HNPCC) stellt die häufigste erbliche Darmkrebsform dar. Verdacht auf HNPCC besteht, wenn mehrere Familienangehörige an Dickdarmkrebs und/oder an Gebärmutterschleimhautkrebs (Endometrium-Karzinom), Dünndarmkrebs, Nierenbecken- oder Harnleiterkrebs erkrankt sind. Ebenfalls verdächtig ist das Auftreten einer dieser Tumoren vor dem 45. Lebensjahr oder wenn ein Patient gleichzeitig oder nacheinander an zwei oder mehreren dieser Tumoren erkrankt.
Ursächlich für HNPCC sind Veränderungen (Mutationen) in den sog. DNA-Reparatur-Genen, die von einer Generation an die nächste weitergegeben werden können. Das Erkrankungsrisiko für das Auftreten eines kolorektalen Karzinoms bis zum 80. Lebensjahr beträgt bei Menschen mit genetischer Prädisposition für HNPCC etwa 80 %.
FAP
Die familiäre adenomatöse Polyposis (FAP) macht etwa 1 % aller kolorektalen Karzinomerkrankungen aus. Die Betroffenen entwickeln hunderte bis tausende kleiner Polypen im Darm. Grundsätzlich kann jedes dieser zunächst gutartigen Adenome zu einem bösartigen Tumor heranwachsen. Die FAP wird durch Veränderungen (Mutationen) im APC-Gen verursacht. Eine solche FAP-Keimbahnmutation kann von einer Generation an die nächste weitergegeben werden. Für Menschen mit einer genetischen Prädisposition für FAP besteht praktisch ein hundertprozentiges Erkrankungsrisiko.
Humangenetische Beratung
Wenn Sie befürchten, dass in Ihrer Familie eine erbliche Veranlagung für Brust- und Eierstockkrebs oder Darmkrebs vorliegt, sollten Sie eine genetische Beratung in Anspruch nehmen. Die Bestimmung des individuellen genetischen Risikos und die Beantwortung der Frage, ob ein erblicher Krebs in der Familie vorliegt, kann nur nach einer ausführlichen Stammbaumanalyse geklärt werden und erfordert humangenetisches Fachwissen. Bei der Stammbaumanalyse müssen die folgenden Faktoren unbedingt erfasst werden:
- Vollständiger Stammbaum über mindestens drei Generationen
- Diagnose aller Tumoren für alle betroffenen Angehörigen
- Alter bei Erstdiagnose für alle Tumorpatienten in der Familie
- Alter und Geschlecht für alle betroffenen und nicht betroffenen Familienangehörigen
Molekulargenetische Diagnostik
Wenn sich der Verdacht auf erblichen Brust- und Eierstockkrebs bzw. HNPCC oder FAP erhärtet, kann eine molekulargenetische Diagnostik sinnvoll sein. Vor Durchführung einer molekulargenetischen Diagnostik werden Sie über die Möglichkeiten und Grenzen der eingesetzten Untersuchungsverfahren, mögliche Ergebnisse und deren Konsequenzen aufgeklärt.
Früherkennung
Spezielle Früherkennungsprogramme und ggf. andere vorbeugende Maßnahmen (z.B. bei erblichem Brustkrebs: medikamentöse Prävention, prophylaktische Operationen) für Personen aus Hochrisikofamilien bzw. mit nachgewiesener pathogener Mutation in einem der derzeit bekannten Krebsgene werden im Beratungsgespräch selbstverständlich auch angesprochen.
Verfasser:
- Dr. Simone Sauter
- FÄ für Humangenetik
- Centrum für Humangenetik Linden
- Kurt-Schumacher-Straße 11
- 35440 Linden
- Telefon: 0 6403 / 68 400
- Telefax: 0 6403 / 68 402
- eMail: sauter@humangenetisches-centrum.de
- eSite: http://www.humangenetisches-centrum.de
Prostatakarzinom
Das Prostatakarzinom ist unter den urologischen Tumoren die häufigste Todesursache und zugleich ab dem 80. Lebensjahr die häufigste tumorbedingte Todesursache überhaupt. Das mittlere Alter bei Diagnose liegt bei 71,4 (SD 8,7) Jahren, das mittlere Sterbealter bei 78 Jahren. 5-Jahresüberleben ist also die Regel und daher geben 5- Jahresüberlebensraten keine Aussage über eine Heilung.
Die jährliche Neuerkrankungsrate je 100.000 Männer steigt mit dem Alter stark an von etwa 50/100.000 bei 60-jährigen bis auf mehr als 400/100.000 bei Lebensalter zwischen 75 und 85 Jahren.
Ätiologie
In letzter Zeit wurde eine familiäre Häufung des Prostatakarzinoms gesichert. Ein Zusammenhang insbesondere mit der Fettaufnahme wird aus ökologischen Studien gefolgert und zeigt deutliche Analogien zum kolorektalen Karzinom und zum Brustkrebs. Andererseits wurden bisher keine Inzidenz-Unterschiede zwischen Stadt und Land nachgewiesen. Schließlich wurden erhöhte Risiken bei Schwermetallexposition, speziell bei Kadmium, gefunden.
Histologie
Epitheliale Tumoren
- I. benigne:
- papilläres Adenom
- II. maligne:
- Adenokarzinom
(kleinazinär, großazinär, kribriform, solide/trabekulär, andere sehr seltene Formen wie z.B. endometroides Karzinom des Utriculus seminalis, papilläres Zystadenokarzinom, muzinöses Karzinom oder adenoid-zystisches Karzinom)
Transitionalzellkarzinom
Plattenepithelkarzinom
Undifferenzierte Karzinome
Kleinzellige Karzinome (selten)
Symptomatik
Im Frühstadium fehlen Symptome völlig, im Spätstadium kommt es zu Miktionsbeschwerden wie bei benigner Prostatahyperplasie (BPH). Dabei können Symptome mit Harnstau bis Urämie vorkommen. Seltener führen auch blutiger Urin oder metastasenbedingte Symptome zur Diagnose eines PC.
Vorsorgeuntersuchungen (Screening)
Der Wert von Vorsorgeuntersuchungen ist umstritten. Das prostataspezifische Antigen (PSA) ist emfindlicher als die digitale rektale Untersuchung
altersabhängige PSA-Werte:
Lebensalter PSA-Wertng/ml
40-49 2,5
50-54 3,555-59 n.n.60-69 4,570-79 6,5
Prognose
Bei einem mittleren Diagnosealter von etwa 72 Jahren und oft sehr langsamem Tumorwachstum sterben viele Patienten an anderen Ursachen, ohne dass ihre Lebensqualität je durch das Prostatakarzinom beeinträchtigt worden wäre. Die Wahl der Therapie muß daher stark von der Lebenserwartung des Patienten, unter Berücksichtigung des Lebensalters und insbesondere der Comorbidität, aber auch von der Aggressivität des Tumors abhängig gemacht werden. Die Überlebenszeit ist abhängig von der Ausdehnung des Tumors und vom Differenzierungsgrad. Ein auf die Prostata begrenztes Karzinom ist oft heilbar und ein medianes Überleben von mehr als 5 Jahren ist die Regel. Patienten mit lokoregionär fortgeschrittenem Tumor, insbesondere mit Lymphknotenmetastasen, sind in der Regel nicht heilbar und ein wesentlicher Teil dieser Patienten wird tumorbedingt sterben, aber das mediane Überleben kann trotzdem im Bereich von 5 Jahren liegen. Bei Fernmetastasierung ist eine Heilung mit den heutigen Therapieformen nicht möglich. Das mediane Überleben liegt bei ein bis drei Jahren und die meisten Patienten sterben am Tumor, aber auch hier kommen langjährige Verläufe ohne wesentlich beeinträchtigte Lebensqualität vor.
Radikale Prostatovesikulektomie
Die radikale Operation ist im allgemeinen nur sinnvoll bei Männern in gutem Gesundheitszustand und Lebensalter unter 70 - 75 Jahren, die einer Operation zustimmen. Die Höhe des PSA korreliert mit dem Ergebnis, so dass ein PSA unter 20 ng/ml als günstig angesehen werden kann (ist aber nicht Bedingung). Vor der Prostatektomie wird in der Regel eine Lymphadenektomie durchgeführt. Diese ist nicht als Therapiemaßnahme anzusehen, sondern dient dazu, dem Patienten die Strapazen einer radikalen Prostatektomie oder Strahlentherapie zu ersparen, falls die Lymphknoten positiv sind.
Strahlentherapie
Bei kleinvolumigem und hochdifferenzierten Tumor mit PSA <20 ng/ml ist die Lymphadenektomie nicht erforderlich, da die Wahrscheinlichkeit von Lymphknotenmetastasen gering ist. Auf eine Beckenbestrahlung wird primär verzichtet. Die Lymphadenektomie kann zur Senkung der Morbidität laparoskopisch durchgeführt werden.
Patienten, die aufgrund internistischer Risikofaktoren für eine radikale Operation nicht in Frage kommen, können mit akzeptabler Nebenwirkungsrate bestrahlt werden, wenn die Bestrahlungstechnik sorgfältig gewählt . Auch eine interstitielle Brachytherapie mit Afterloading-Technik oder die interstitielle Implantation radioaktiver Strahler wurde in einer Reihe mit guten Ergebnissen untersucht.
Hormontherapie
Das Prostatacarcinom ist eine Tumorart, bei der eine Behandlung mir Hormonen neben den Standardtherapien Strahlentherapie und Operation sehr erfolgversprechend sind. Die Hormontherapie wird entweder in Kombination mit anderen Verfahren eingesetzt (adjuvante Therapie) oder aber auch alleine, hier oft bei fortgeschrittenen Tumoren, bei denen keine endgültige Heilung mehr erreichbar ist. In diesen Fällen führt die Hormonbehandlung zu Verbesserung von Krankheitssymptomen und Verlängerung des Überlebens. Das Prinzip der Therapie besteht in einer möglichst kompletten Ausschaltung der männlichen Geschlechthormone. Hierzu kann eine Entfernung der Hoden (Orchiektomie) vorgenommen werden oder aber durch Beeinflussung der Androgenproduktion die Hormonproduktion chemisch ausgeschaltet werden (LH-RH Agonisten). Zusätzlich können weitere Stoffe zu einer maximalen Hormonsuppression nötig sein (z.B.Bicalutamid etc.)
Chemotherapie
Die Chemotherapie des fortgeschrittenen, endokrin-resistenten Prostata- Karzinoms ist weiterhin unbefriedigend. Dennoch könne mit den heute zur Verfügung stehenden Medikamenten oft gute leidenslindernde Therapie durchgeführt werden. Besondere Erwähnung verdient hier das Medikament Estramustin, mit dem sich oft noch andauernde Remissionen erreichen lassen.
Verfasser:
- Dr. med. Gerson Lüdecke
- Facharzt für Urologie
- Leitender Oberarzt am Universitätsklinikum Gießen -
- Klinik und Poliklinik für Urologie und Kinderurologie
- Rudolf-Buchheim-Straße 735385 Gießen
- Telefon: 0641 99 45 260
- Telefax: 0641 99 45 259
- eMail: gerson.luedecke@email.de
-
Ovarialkarzinom
Bösartige Tumore, die von den Eierstöcken ausgehen. Das Risiko, an einem Ovarialkarzinom zu erkranken, steigt mit dem Alter an, das Durchschnittsalter liegt bei ca. 60 Jahren. Aber insbesondere bei bestimmten Tumortypen können auch junge Frauen betroffen sein.
Risikofaktoren
Der wichtigste Risikofaktor ist die familiäre Disposition (Neigung) zu dieser Erkrankung. Genetisch besteht ein Zusammenhang zwischen dem Ovarialkarzinom und dem Brustkrebs (Mammakarzinom), sodaß auch Brustkrebsneigung in der Familie das Risiko erhöht. Offenbar spielt die Anzahl der stattgehabten Eisprünge im Leben der Frau eine Rolle, denn nicht nur Schwangerschaften (während denen natürlich kein Eisprung stattfindet), sondern auch die langfristige Einnahme der Pille (die den Eisprung verhindert – Ovulationshemmer) senken das Risiko!
Symptome
Da die Eierstöcke viel Platz im Bauchraum und keine Verbindung über die Scheide nach aussen haben, sind Ovarialkarzinome oft lange symptomlos. Daher werden sie meist spät erkannt, was die Prognose (Aussicht auf Heilung) verschlechtert. Da sich in fortgeschrittenen Stadien meist Wasser im Bauch (Aszites) ansammelt, fällt die Krankheit häufig nur durch eine Bachumfangszunahme, Verdauungsbeschwerden oder allgemeine Schwäche auf. Gelegentlich können Blutungen auftreten. Durch eine vaginale Ultraschalluntersuchung im Rahmen der Krebsvorsorge können Ovarialkarzinome zufällig in frühen Stadien, die mit viel höherer Heilungsaussicht behandelt werden können, festgestellt werden. Die vaginale Ultraschalluntersuchung ist leider kein Bestandteil der Krebsvorsorge und muß meist privat bezahlt werden.
Diagnostik
Neben der gynäkologischen Tastuntersuchung ist die vaginale und abdominale (vom Bauch her) Ultraschalluntersuchung die wichtigste Untersuchung. Bei grösseren Tumoren ist die Darmspiegelung und die Computertomographie (Schichtröntgen) notwendig, um die Ausbreitung des Tumors zu beurteilen und die Operation vorzubereiten.
Therapie
Die operative Behandlung des Ovarialkarzinoms besteht aus der Entfernung der Gebärmutter und der Eierstöcke sowie des grossen Netzes, eines am Darm anhängenden Lymphorganes des Bauchraumes, über einen Bauchschnitt. Ziel ist die vollständige, falls nicht möglich die weitgehende Entfernung des Tumorgewebes. Daher ist in vielen Fällen die Entfernung von Darmanteilen und die Ausräumung der Lymphknoten an den Blutgefässen des Bauches und Beckens notwendig. Die Operation wird bei Darmbeteiligung interdisziplinär (gemeinsam von Gynäkologen und Viszeralchirurgen) durchgeführt. Da Ovarialkarzinome sehr chemotherapieempfindlich sind, wird nach der Operation fast immer eine Chemotherapie durchgeführt.
Risikofaktoren
Der wichtigste Risikofaktor ist die familiäre Disposition (Neigung) zu dieser Erkrankung. Genetisch besteht ein Zusammenhang zwischen dem Ovarialkarzinom und dem Brustkrebs (Mammakarzinom), sodaß auch Brustkrebsneigung in der Familie das Risiko erhöht. Offenbar spielt die Anzahl der stattgehabten Eisprünge im Leben der Frau eine Rolle, denn nicht nur Schwangerschaften (während denen natürlich kein Eisprung stattfindet), sondern auch die langfristige Einnahme der Pille (die den Eisprung verhindert – Ovulationshemmer) senken das Risiko!
Symptome
Da die Eierstöcke viel Platz im Bauchraum und keine Verbindung über die Scheide nach aussen haben, sind Ovarialkarzinome oft lange symptomlos. Daher werden sie meist spät erkannt, was die Prognose (Aussicht auf Heilung) verschlechtert. Da sich in fortgeschrittenen Stadien meist Wasser im Bauch (Aszites) ansammelt, fällt die Krankheit häufig nur durch eine Bachumfangszunahme, Verdauungsbeschwerden oder allgemeine Schwäche auf. Gelegentlich können Blutungen auftreten. Durch eine vaginale Ultraschalluntersuchung im Rahmen der Krebsvorsorge können Ovarialkarzinome zufällig in frühen Stadien, die mit viel höherer Heilungsaussicht behandelt werden können, festgestellt werden. Die vaginale Ultraschalluntersuchung ist leider kein Bestandteil der Krebsvorsorge und muß meist privat bezahlt werden.
Diagnostik
Neben der gynäkologischen Tastuntersuchung ist die vaginale und abdominale (vom Bauch her) Ultraschalluntersuchung die wichtigste Untersuchung. Bei grösseren Tumoren ist die Darmspiegelung und die Computertomographie (Schichtröntgen) notwendig, um die Ausbreitung des Tumors zu beurteilen und die Operation vorzubereiten.
Therapie
Die operative Behandlung des Ovarialkarzinoms besteht aus der Entfernung der Gebärmutter und der Eierstöcke sowie des grossen Netzes, eines am Darm anhängenden Lymphorganes des Bauchraumes, über einen Bauchschnitt. Ziel ist die vollständige, falls nicht möglich die weitgehende Entfernung des Tumorgewebes. Daher ist in vielen Fällen die Entfernung von Darmanteilen und die Ausräumung der Lymphknoten an den Blutgefässen des Bauches und Beckens notwendig. Die Operation wird bei Darmbeteiligung interdisziplinär (gemeinsam von Gynäkologen und Viszeralchirurgen) durchgeführt. Da Ovarialkarzinome sehr chemotherapieempfindlich sind, wird nach der Operation fast immer eine Chemotherapie durchgeführt.
Verfasserin:
- Roswitha Mersmann
- ehem. leitende Oberärztin der Gynäkologischen Abteilung
- Asklepios-Klinik Lich
- Goethestraße 4
35423 Lich
Telefon: 06404 81 0
Cervixkarzinom (Gebärmutterhalskrebs)
Bösartige Tumore, die vom Gebärmutterhals – dem Teil der Gebärmutter, der in die Scheide hineinragt – ausgehen. In diesem Bereich grenzt das Zylinderepithel, das die Gebärmutter auskleidet, an das die Scheide auskleidende Plattenepithel. Beide Zellarten konkurrieren durch verstärktes Wachstum um diese Gewebsgrenze. 90 % der Zervixkarzinome gehen vom Plattenepithel, 10 % vom Zylinderepithel aus. Zervixkarzinome kommen in allen Altersgruppen vor, betreffen aber häufig auch junge Frauen (30.-40.LJ).
Risikofaktoren
Da bestimmte, sexuell übertragbare Viren (HPV =humane Papilloma Viren) in der Entstehung dieser Karzinome maßgeblich beteiligt sind, ist das Sexualverhalten ein wichtiger Risikofaktor, insbesondere ein häufiger Partnerwechsel. Auch das Rauchen erhöht das Risiko stark, da sich die krebserregenden Stoffe der Zigarette im Gebärmutterhalsschleim anreichern.
Symptome
Die Krebsvorstufen und kleinere Tumore sind oft symptomarm, sie machen sich gelegentlich durch Kontaktblutungen (Blutungen während oder nach dem Geschlechtsverkehr) bemerkbar. Größere Tumore verursachen einen verstärkten, meist fleischwasserartigen Ausfluß und unregelmässige Blutungen.
Diagnostik
Der Gebärmutterhalskrebs ist die gynäkologische Tumorart, die der Vorsorge am besten zugänglich ist, da direkt „vor Ort“ der Vorsorgeabstrich entnommen wird. Bei Frauen, die regelmässig ab dem 3. Lebensjahrzehnt zur Krebsvorsorge gehen, treten diese Tumore nur 10 x seltener auf als bei anderen Frauen. Neben der gynäkologischen Untersuchung, der Abstrichentnahme und der Kolposkopie (Betrachtung des Muttermundes mit dem Mikroskop), mit denen man bereits Krebsvorstufen sehr gut nachweisen kann, ist auch eine Untersuchung des Gewebes auf HPV – Viren möglich. Unter Einsatz dieser Methoden kann man diese Tumore fast immer in Vor- oder Frühstadien erkennen, bevor sie der betreffenden Frau gefährlich werden. Wenn der Abstrich suspekt (nicht in Ordnung) ist, wird im Krankenhaus eine Konisation (Gewebeentnahme) durchgeführt. Hierdurch wird sowohl eine genaue feingewebliche Diagnose als auch die Entfernung des erkrankten Bezirkes möglich.
Therapie
In Vorstadien ist die Konisation ausreichend, in den frühen Stadien, in denen der Tumor auf den Gebärmutterhals und die direkte Umgebung begrenzt ist, wird die Gebärmutter mitsamt dem bindegewebigen Halteapparat bis zu den Beckenwänden radikal entfernt. In fortgeschrittenen Stadien kommt die Bestrahlung und eine lokale (örtliche) oder systemische (im ganzen Körper wirkende) Chemotherapie zum Einsatz.
Risikofaktoren
Da bestimmte, sexuell übertragbare Viren (HPV =humane Papilloma Viren) in der Entstehung dieser Karzinome maßgeblich beteiligt sind, ist das Sexualverhalten ein wichtiger Risikofaktor, insbesondere ein häufiger Partnerwechsel. Auch das Rauchen erhöht das Risiko stark, da sich die krebserregenden Stoffe der Zigarette im Gebärmutterhalsschleim anreichern.
Symptome
Die Krebsvorstufen und kleinere Tumore sind oft symptomarm, sie machen sich gelegentlich durch Kontaktblutungen (Blutungen während oder nach dem Geschlechtsverkehr) bemerkbar. Größere Tumore verursachen einen verstärkten, meist fleischwasserartigen Ausfluß und unregelmässige Blutungen.
Diagnostik
Der Gebärmutterhalskrebs ist die gynäkologische Tumorart, die der Vorsorge am besten zugänglich ist, da direkt „vor Ort“ der Vorsorgeabstrich entnommen wird. Bei Frauen, die regelmässig ab dem 3. Lebensjahrzehnt zur Krebsvorsorge gehen, treten diese Tumore nur 10 x seltener auf als bei anderen Frauen. Neben der gynäkologischen Untersuchung, der Abstrichentnahme und der Kolposkopie (Betrachtung des Muttermundes mit dem Mikroskop), mit denen man bereits Krebsvorstufen sehr gut nachweisen kann, ist auch eine Untersuchung des Gewebes auf HPV – Viren möglich. Unter Einsatz dieser Methoden kann man diese Tumore fast immer in Vor- oder Frühstadien erkennen, bevor sie der betreffenden Frau gefährlich werden. Wenn der Abstrich suspekt (nicht in Ordnung) ist, wird im Krankenhaus eine Konisation (Gewebeentnahme) durchgeführt. Hierdurch wird sowohl eine genaue feingewebliche Diagnose als auch die Entfernung des erkrankten Bezirkes möglich.
Therapie
In Vorstadien ist die Konisation ausreichend, in den frühen Stadien, in denen der Tumor auf den Gebärmutterhals und die direkte Umgebung begrenzt ist, wird die Gebärmutter mitsamt dem bindegewebigen Halteapparat bis zu den Beckenwänden radikal entfernt. In fortgeschrittenen Stadien kommt die Bestrahlung und eine lokale (örtliche) oder systemische (im ganzen Körper wirkende) Chemotherapie zum Einsatz.
Verfasserin:
- Roswitha Mersmann
- ehem. leitende Oberärztin der Gynäkologischen Abteilung
- Asklepios-Klinik Lich
- Goethestraße 4
35423 Lich
Telefon: 06404 81 0
Vaginalkarzinom (Scheidenkrebs)
Bösartige Tumore, die von der Scheidenwand (Vagina) ausgehen. Dieser Tumor ist im Vergleich zu den anderen gynäkologischen Tumoren sehr selten. Das Risiko, an einem Vaginalkarzinom zu erkranken, steigt mit dem Alter an.
Risikofaktoren
Da bestimmte, sexuell übertragbare Viren (HPV =humane Papilloma Viren) in der Entstehung dieser Karzinome maßgeblich beteiligt sind, ist das Sexualverhalten ein Risikofaktor, insbesondere ein häufiger Partnerwechsel. Bei Diabetikerinnen ist das Erkrankungsrisiko, bedingt durch die Neigung zu chronischen Infektionen im Genitalbereich, erhöht.
Symptome
Die wichtigsten Symptome sind Ausfluß und vaginale Blutungen, die spontan (ohne äussere Einflüsse) oder nach Geschlechtsverkehr auftreten können. Auch Schmerzen beim Wasserlassen oder bei der Darmentleerung können bestehen. Da das Vulvakarzinom, das Vaginalkarzinom und das Ovarialkarzinom auch nach Entfernung der Gebärmutter auftreten können, ist auch nach Entfernung der Gebärmutter die gynäkologische Krebsvorsorgeuntersuchung wichtig.
Diagnostik
Das Vaginalkarzinom ist bei der gyn. Vorsorgeuntersuchung erkennbar, im Zweifelsfall kann ein Vorsorgeabstrich entnommen werden oder sogar eine Gewebsprobe. Neben der gynäkologischen Untersuchung, der Abstrichentnahme und der Kolposkopie (Betrachtung der Scheidenwand mit dem Mikroskop), mit denen man bereits Krebsvorstufen nachweisen kann, ist auch eine Untersuchung des Gewebes auf HPV – Viren möglich.
Therapie
In Vor- und Frühstadien ist eine operative Entfernung der betroffenen Areale ausreichend. Bei grösseren Tumoren wird die gesamte Scheide einschließlich der Lymphknoten in Leiste und Becken operativ entfernt. In fortgeschrittenen Stadien kommt die Bestrahlung, in seltenen Fällen eine Chemotherapie zum Einsatz
Risikofaktoren
Da bestimmte, sexuell übertragbare Viren (HPV =humane Papilloma Viren) in der Entstehung dieser Karzinome maßgeblich beteiligt sind, ist das Sexualverhalten ein Risikofaktor, insbesondere ein häufiger Partnerwechsel. Bei Diabetikerinnen ist das Erkrankungsrisiko, bedingt durch die Neigung zu chronischen Infektionen im Genitalbereich, erhöht.
Symptome
Die wichtigsten Symptome sind Ausfluß und vaginale Blutungen, die spontan (ohne äussere Einflüsse) oder nach Geschlechtsverkehr auftreten können. Auch Schmerzen beim Wasserlassen oder bei der Darmentleerung können bestehen. Da das Vulvakarzinom, das Vaginalkarzinom und das Ovarialkarzinom auch nach Entfernung der Gebärmutter auftreten können, ist auch nach Entfernung der Gebärmutter die gynäkologische Krebsvorsorgeuntersuchung wichtig.
Diagnostik
Das Vaginalkarzinom ist bei der gyn. Vorsorgeuntersuchung erkennbar, im Zweifelsfall kann ein Vorsorgeabstrich entnommen werden oder sogar eine Gewebsprobe. Neben der gynäkologischen Untersuchung, der Abstrichentnahme und der Kolposkopie (Betrachtung der Scheidenwand mit dem Mikroskop), mit denen man bereits Krebsvorstufen nachweisen kann, ist auch eine Untersuchung des Gewebes auf HPV – Viren möglich.
Therapie
In Vor- und Frühstadien ist eine operative Entfernung der betroffenen Areale ausreichend. Bei grösseren Tumoren wird die gesamte Scheide einschließlich der Lymphknoten in Leiste und Becken operativ entfernt. In fortgeschrittenen Stadien kommt die Bestrahlung, in seltenen Fällen eine Chemotherapie zum Einsatz
Verfasserin:
- Roswitha Mersmann
- ehem. leitende Oberärztin der Gynäkologischen Abteilung
- Asklepios-Klinik Lich
- Goethestraße 4
35423 Lich
Telefon: 06404 81 0
Malignes Melanom
Definition
Maligner (bösartiger ) Tumor, der vom
melanozytären (melaninbildenden) Zellsystem ausgeht und überwiegend an
der Haut, seltener auch an Schleimhäuten, am Auge oder an den Hirnhäuten
auftritt. Das Melanom ist zumeist stark pigmentiert, selten
amelanotisch (hautfarben). 90 % der Todesfälle an Hautkrebs sind dem
Malignen Melanom zuzuordnen. Es besteht eine frühe Tendenz zur
Metastasierung vorrangig in Lymphgefäße aber auch Blutgefäße. Man
unterscheidet vier Melanom-Typen, die sich durch Aussehen und
histologische (feingewebliche) Untersuchung unterscheiden (Superfiziell
spreitendes Melanom, Noduläres Melanom, Lentigo maligna Melanom, Akral
lentiginöses Melanom).
Epidemiologie
Bei der weißen Bevölkerung nehmen
Melanome weltweit zu. In Mitteleuropa kommen diese bei 10-12 Fällen pro
100.000 Einwohnern und Jahr vor. Die Verteilung von Männern zu Frauen
liegt bei 1:1,5. Das Auftreten ist gehäuft zwischen 30 und 70 Jahren mit
Zunahme im hohen Alter.
Risikofaktoren
Exzessive Sonnenbestrahlung bis zum
20. Lebensjahr mit Sonnenbränden fördert das Auftreten. Genetische
Veranlagung ist ein wichtiger Risikofaktor; 5-10% der Melanome treten in
erblich belasteten Familien auf. Weiterhin sind Patienten mit vielen
Nävi (Hautmalen), atypischen Nävi und hellem Hauttyp extrem gefährdet.
Melanome entstehen zu 2/3 neu, zu 1/3 auf vorbestehenden Nävi.
Symptome
Das Melanom ist anfangs in der Regel
symptomlos. Fortgeschrittene Melanome fallen zeitweilig durch Bluten,
Geschwüre oder Größenzuwachs auf. Früherkennung und rechtzeitige
Therapie können durch jährlich durchgeführte „Melanomvorsorgen“ beim
Dermatologen erreicht werden.
Diagnostik
Bei der „Melanomvorsorge“ untersucht
der Dermatologe den Patienten von Kopf bis Fuß mit einem Dermatoskop
(Hautmikroskop). Bei der Verdachtsdiagnose eines Melanoms folgt die
Operation mit histologischer Untersuchung. Bestätigt sich der Verdacht,
erfolgen weitere diagnostische Verfahren wie Lymphknotenbiopsie und
bildgebende Verfahren. Regelmäßige Blutuntersuchungen mit Bestimmung von
Tumormarkern sind ebenfalls erforderlich. Der Patient stellt sich nun
in kürzeren Zeitabständen zur „Melanomnachsorge“ vor.
Therapie
Je nach Tumorstadium des Melanoms
erfolgt die Exzision (operative Entfernung) mit einem Sicherheitsabstand
zwischen 0,5cm und 2cm. Bei Verdacht auf Lymphknotenmetastasen ist
evtl. eine „Schildwächterlymphknotenbiopsie“(Wächterlymphknoten:der
Lymphknoten einer regionären Lymphknotenstation, der direkten Zufluss
vom drainierten Lymphgefäß hat) mit Lymphabstromszintigraphie aus dem
Tumorgebiet erforderlich. Bei Lymphknotenmetastasierung folgt in der
Regel die radikale Lymphknoten-Dissektion (Entfernung) evtl. mit
zusätzlicher Immuntherapie.
Als alternative Methode kommt die
Strahlentherapie bei älteren Patienten mit hohem Operationsrisiko in
Frage. Hautmetastasen werden üblicherweise chirurgisch, durch
Kryotherapie (Vereisung)) oder Lasern entfernt. Knochen- und
Hirnmetastasen können mit Strahlentherapie evtl. zusätzlich
Chemotherapie, Fernmetastasen mit Immuntherapie und Chemotherapie
behandelt werden. Kontrolluntersuchungen erfolgen je nach Tumordicke und
Stadium in drei- bis sechsmonatlichen Abständen über 10 Jahre.
Plattenepithelkarzinom
Synonyme: Spinaliom, Spinozelluläres Karzinom, Stachelzellkarzinom
Definition
Maligner Tumor der Epidermis mit
unterschiedlich schnellem Wachstum, der im Verlauf in tiefere
Hautschichten und Nachbargewebe wächst. Er metastasiert zumeist in
Lymphgefäße.
Epidemiologie
Häufiger Tumor mit zunehmendem
Auftreten (Männer 100/100000/Jahr, Frauen 50/100000/Jahr). An den
Haut-Schleimhaut-Übergängen häufigster bösartiger Tumor. Das
Haupterkrankungsalter liegt zwischen dem 60. und 80. Lebensjahr.
Risikofaktoren
Chronische Lichtexposition,
Röntgenbestrahlung, Chemokanzerogene (Teer, Arsen), Immunsuppression
(AIDS,Organtransplantation), selten Entstehung auf
chronisch-entzündlicher Haut oder Narben.
Diagnostik
Bereits beim Auftreten einer
„Aktinischen Keratose“ (erstes Stadium eines Plattenepithelkarzinoms)
mit Schuppen- oder Krustenbildung auf lichtexponierter Haut sollte
sofort der Hautarzt aufgesucht werden.
Therapie
Im Anfangsstadium der Aktinischen
Keratose ist häufig noch Behandlung mit spezifischen Cremes, Stickstoff
oder Kürettage (Chirurgisches Abtragen) möglich. Das
Plattenepithelkarzinom sollte komplett exzidiert und histologisch
kontrolliert werden. Nur bei Inoperabilität kommen alternativ
Röntgenbestrahlung, Kryo- und Lasertherapie in Frage.
Basaliom
Synonyme. Basalzellkarzinom, Basalzellenkrebs, Epithelioma basocellulare, Trichoblastisches Karzinom
Definition
Maligner, epithelialer Tumor mit lokal invasivem Wachstum aber ohne Metastasierungstendenz.
Epidemiologie
Häufigster maligner Tumor der Haut
(Männer 200/100000, Frauen 100/100000/Jahr) mit steigender Häufigkeit.
Es entwickelt sich aus entarteten Zellen der tiefsten Schicht der
Oberhaut, der sogenannten Basalzellschicht. Das Basaliom tritt vorrangig
bei älteren Personen auf.
Risikofaktoren
Chronische Lichtexposition, chemische
Kanzerogene (Arsen), Immunsuppression, genetische Prädisposition (helle
und lichtempfindliche Hauttypen, Basalzellnävussyndrom),
Strahlentherapie.
Symptome
Hauptlokalisation ist das Gesicht.
Treten Basaliome gehäuft auf, finden sie sich bevorzugt am Stamm. Das
initiale Basaliom sieht klinisch sehr unterschiedlich aus. Das Wachstum
ist langsam. Oft zeigt sich zu Beginn eine kaum linsengroße grauweiße
Verhärtung mit winzigen Gefäßen. Durch Kratzen oder Rasieren entsteht
nicht selten eine immer an der gleichen Stelle auftretende Blutkruste.
Diagnostik
Untersuchung mit dem Dermatoskop (Hautmikroskop) durch den Hautarzt.
Therapie
Bei Basaliomverdacht folgt die
Exzision mit histologischer Randkontrolle. Nur selten z.B. bei älteren
Patienten mit ausgedehntem oberflächlichem Basaliom oder bei schwierigen
Lokalisationen kommt die Strahlentherapie in Frage. Im Anfangsstadium
kommen Kürrettage, Kryotherapie und spezifische Cremes (unter ärztlicher
Kontrolle) als Therapieoption in Betracht.
Verfasser/in:
- Dr. med. Birgit Uhl-Pelzer
Fachärztin für Haut- und Geschlechtskrankheiten
Hungener Straße 12
35423 Lich
Telefon: 06404-950405
eMail: dr.uhl-pelzer@t-online.de
eSite: http://www.uhl-pelzer.de
Bronchialkarzinom | mediastinale Tumore | Pleuramesotheliom
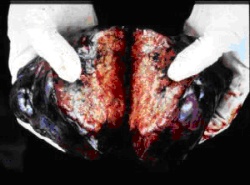
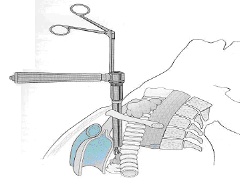
Weitere Operationsverfahren sind:
____________________________________________________
Bronchialkarzinom (sog. Lungenkrebs)
Das Bronchialkarzinom ist mit einer Inzidenz (Häufigkeit) von ca. 38.100 Neuerkrankungen pro Jahr der am häufigsten zum Tode führenden maligne (bösartiger) Tumor weltweit. Die Neuerkrankungsrate ist beim Mann ca. 60-70 Erkrankungen auf 100.000 Männer. Position 1 der tumorbedingten Todesfälle. Dagegen nur 7-9 Neuerkrankungen auf 100.000 Frauen. Bei der Frau der dritthäufigster Tumor (8%), hinter Malignomen der Brust und des Dickdarm (Kolon), Tendenz steigend. Männer : Frauen wie 5:1. Der Altersgipfel liegt in der 6. Lebensdekade. Es besteht kein Zweifel daran, dass das Rauchen der bedeutendste Einzelrisikofaktor für das Bronchialkarzinom ist. Unter den beruflichen Noxen (Giften) ist das Asbest anerkannt.

Es werden grundsätzlich zwei Tumorzellarten unterschieden. Die größte Gruppe stellt mit 75-80% der Patienten das nicht-kleinzellige Bronchialkarzinom (NSCLC) dar. Bei ca. 20-25% der Patienten wird ein prognostisch ungünstigeres kleinzelliges Bronchialkarzinom (SCLC) diagnostiziert. Die spätere Metastasierungstendenz, die niedrigere Zellteilungsrate und die langsamere Wachstumsgeschwindigkeit unterscheidet das nicht–kleinzellige vom kleinzelligen Bronchialkarzinom.
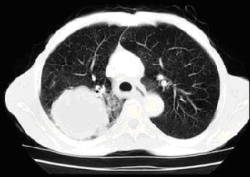 

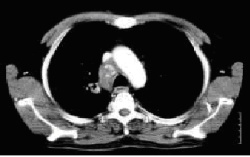
 Abb.1: Großes Bronchialkarzinom im Thorax-CT
Abb.2: Operationspräparat mit demselben Bronchialkarzinom (rechter Lungenoberlappen)
Nach der Verdachtsdiagnose eines Bronchialkarzinoms (meist in der Röntgen-Aufnahme der Lunge) durch den behandelnden Arzt (Hausarzt, Internist, Lungenfacharzt) wird durch weitere Bildgebung und mögl. Gewebeentnahme die lokale Tumorausdehnung sowie der Ausschluss/Nachweis einer Fernmetastasierung (Tumorstreuung) bestimmt.
Folgende Untersuchungen sind zur Einstufung des Ausmaß der Tumorerkrankung (Staging) und Therapieentscheidung (Operation u./o. Chemotherapie) in der Zusammenschau notwendig:
- Computertomographie des Brustkorbes
- (Thorax-CT; siehe Abb. 1)
- Oberbauch-Sonografie (Ultraschall Untersuchung)
- Oberbauch–CT
- Lungenfunktion
- Bronchoskopie (Bronchienspiegelung)
Ergänzend könnte zusätzlich notwendig sein:
Spezielle Computertomographie mit nuklearem. Marker (PET-CT)
Skelettszintigraphie (spezielle Knochenstoffwechsel- Untersuchung)
Computertomographie des Kopfes (CCT)
Herzecho, Untersuchung der Herzfunktion in Ruhe (ggf. auch mit Belastung)
Abb. 3: Bronchialkarzinom im rechten Lungenoberlappen, Befund bei der Bronchoskopie
Als operative Untersuchungsverfahren können darüberhinaus noch folgende Maßnahmen vor der endgültigen Therapiefestlegung erforderlich sein:
- Mediastinoskopie (Mittelfellspiegelung)
- Thorakoskopie, (Brustkorbspiegelung)
- Gewebeentnahme von auffälligen Hals-Lymphknoten (Tochtergeschwülste ?)
Abb: Mediastinoskopie, schematisch
Anhand der Untersuchungsbefunde aus Bildgebung und Gewebeuntersuchung wird das Tumorstadium (nach UICC: Stadium I-IV) und die daraus abzuleitende individuelle Therapie des Patienten festgelegt. Voraussetzung für eine optimale Behandlung und eine möglichst günstige Prognose der Erkrankung ist das frühzeitige Erkennen und ein frühes Erkrankungsstadium.
Für die Prognose des Patienten ist die Art des Lungentumors (Entität), die Tumorausbreitung und die Möglichkeit der kompletten chirurgischen Resektion die drei wichtigsten Faktoren. Ganz entscheidend ist bei den o.g. Untersuchungen hierbei, ob der Tumor:
- lokal auf die Lunge begrenzt ist
- lokal fortgeschritten und in die Nachbarorganstrukturen eingewachsen ist oder mediastinale Lymphknoten (Lymphknoten des Mittelfellraumes) gestreut hat oder
- bereits jenseits der Lunge gestreut ist (metastasiert ist)
Am Ende der Diagnostik sind im Sinne der individuell bestmöglichen und sinnvollen Behandlung grundsätzlich zwei verschiedene Therapieziele möglich.
- kurativer Therapieansatz (mit dem Ziel einer definitiven Heilung) oder
- palliativer Therapieansatz (Verlängerung der Lebenszeit, Abmilderung der Tumorsymptome und Besserung der Lebensqualität).
Allerdings liegt bei Diagnosestellung des Bronchialkarzinoms in ca. 30% der Fälle ein lokal fortgeschrittener Tumor vor (mit mediastinalen Lymphknotenmetastasen oder Tumorinfiltration in benachbarte Gefäß- oder Organstrukturen wie z.B. Brustwand, Herzbeutel, Zwerchfell, obere Hohlvene oder Aorta; Tumor-Stadium IIIb in der Klassifikation nach UICC) und bei weiteren 40% ein bereits fern-metastasierter Tumor (Metastasen / Tochterabsiedlungen in anderen Organen oder Lymphknoten außerhalb des Thorax Brustkorb); Stadium IV in der Klassifikation nach UICC. Beim Stadium IV ist eine Heilung nicht möglich.
Therapiekonzept – Operationsplanung:
Ein lokal begrenztes oder im Frühstadium nachgewiesenes Bronchialkarzinom (Stadium I+II und IIIa (wenn T3 N1+N2)) sollte durch eine thoraxchirurgische Operation behandelt werden. Hier besteht ein kurativer Behandlungsansatz. Dabei wird der tumorerkrankte Lungenabschnitt und zusätzlich immer die gesamten Lymphknoten im Bereich der erkrankten Thoraxseite entfernt.
Im Falle eines lokal fortgeschrittenen Bronchialkarzinoms (Stadium III), sowie bei intraoperativem Nachweis befallener Lymphknoten ist neben der Operation zusätzlich eine Chemotherapie und/oder Strahlentherapie erforderlich. Im Stadium IIIb (keine Fernmetastase) wird fallbezogen eine Chemotherapie (Induktionsbehandlung) vor der Operation empfohlen. Ist eine Operation auf Grund einer Fernmetastasierung (Stadium IV), weiterer Erkrankungen bzw. wegen erheblich reduzierter Lungenfunktion dem Patienten nicht zuzumuten, wird durch den Onkologen eine Zweifach-Chemotherapie (ggf. in Kombination mit platinhaltigen Substanzen) durchgeführt.
Wichtig ist, dass in einer gemeinsamen ärztlichen Konferenz unter Beteiligung eines Thoraxchirurgen, Pneumologen (Lungenfacharzt), internistischen Onkologen (Tumorspezialist) und Strahlentherapeuten wird das jeweilige Therapiekonzept für jeden Patienten individuell besprochen und festgesetzt.
Grundsätzlich sind 5 Behandlungsstrategien möglich:
- alleinige Operation
- präoperative Strahlentherapie + Operation (z.B. bei sog. Pancoast-Tumor, siehe unten)
- präoperative Chemotherapie + Operation (Induktionsbehandlung, neoadjuvant)
- Operation + postoperative Chemo-/Strahlentherapie
- alleinige Chemo-- u./o. Strahlentherapie
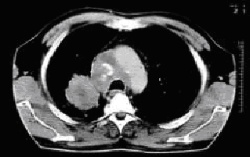
Abb. 4: Verkleinerung eines Bronchialkarzinoms durch präoperative (neoadjuvante) Chemotherapie (Thorax-CT im Verlauf, oben vor der Chemotherapie, unten nach der Chemotherapie)
Vor der Operation werden die Patienten intensiv krankengymnastisch (insbes. in der Atemtherapie) geschult, damit das Risiko der Operation möglichst gering gehalten werden kann. Ein Beenden des Rauchens vor der Operation muss unbedingt angestrebt werden.
Standard-Operationsverfahren sind:
- Lobektomie (Entfernung eines Lungenlappen, s. Abb. 2)
- Pneumektomie (Entfernung eines Lungenflügels)
Diese sind die klassischen Resektionsverfahren. Dehnt sich der Tumor oder die entzündliche Infiltration (Begleitreaktion) bis zur Pleura (Rippenfellhaut) aus, muss dieser Lungeabschnitt über diesen Bereichen außerhalb der Rippenhaut (extrapleural) ausgelöst werden. Man spricht hier noch nicht von einer erweiterten Resektion.
Keil- und Segmentresektionen sind in der Regel zur Entfernung von Tochtergeschwülsten (Metastasen) indiziert.Erweiterte Resektionsverfahren, wie z.B. die intraperikardiale Gefäßversorgung, Vorhofteilresektion, Resektion der Brustwand, des Zwerchfells oder von Ösophaguswandanteilen en-bloc mit Entfernung des Primärtumors können notwendig sein, um eine komplette Entfernung im Gesunden zu erzielen. Grundsätzlich muss immer mit ausreichendem Abstand zum Tumor, in Korrespondenz zur Tumorausbreitung oder dem Lymphknotenbefall, gegebenenfalls in Abhängigkeit von der Schnellschnittuntersuchung (Resektionslinie am Bronchus, Pleura, Gefäß), operiert werden.Parenchym sparende Resektionsverfahren (Manschettenlobektomie), Plastische Eingriffe an der Trachea, Bronchien und Gefäßen ermöglichen bei entsprechenden Tumorstadien ein gewebesparendes und funktionserhaltendes Operieren. Diese gilt nicht nur für die sogenannten Manschettenresektionen, sondern für alle Eingriffe einschließlich der Bifurkationsresektion (Aufteilung der Hauptluftröhre in den rechten+ linken Hauptbronchus) und End-zu-End-Anastomose des gegenseitigen Hauptbronchus mit der Trachea (Hauptluftröhre).
Endoskopische Verfahren (minimal-invasiv, Schlüssellochtechnik) sind bei Resektionen von Bronchialkarzinomen Gegenstand der Forschung und sind zurzeit kein anerkannter Standard. Sog. Lungen-Rundherde d.h. Lungenherde unklaren Verhaltens (benigne/maligne) können oft zunächst endoskopisch entnommen werden. Bei Malignität folgt dann aber immer mindestens die o.g. Lappenentfernung über einen herkömmlichen Rippenrandschnitt.
Bei den sog. Pancoast-Tumoren in der oberen Thoraxapertur (Bronchialkarzinom, ausgehend vom Oberlappen mit Anwachsen an die Rippen der oberen Thoraxöffnung) handelt es sich je nach Lymphknotenbefall im Mediastinum (Mittelfell) um einen lokal fortgeschrittenen, aber potentiell resektablen Tumor (TNM: T3 oder T4, Stadium IIIa oder IIIb). Hier ist eine Vorbestrahlung mit 50-60 Gray und ggf. eine Chemotherapie vor Operation erforderlich. Die Resektion (gegebenenfalls mit Rippenresektion kombiniert) muss 12-14 Tage nach Beendigung der Bestrahlung erfolgen.
Prognose: Die Überlebenschance nach vollständiger (kurativer) Resektion eines Bronchialkarzinoms ist vom Tumor Stadium abhängig und korreliert vor allem mit dem Vorhandensein und Ausdehnung des Lymphknotenbefalles. Im Frühstadium I (nach UICC) werden 5-Jahres-Überlebensraten bis zu 80- 90% erreicht. Die durchschnittlichen 5-Jahres-Überlebensraten nach Resektion für die Tumorstadien II und IIIA (nach UICC) liegen bei 40 % bzw. 20-25%. Auch bei lokal fortgeschrittenen Tumoren können günstige Ergebnisse erzielt werden. Bei Brustwand- und Zwerchfellbefall oder Tumorausdehnung bis zur Aufteilung der Luftröhre (Bifurkation) werden Überlebensraten von 23-42% mitgeteilt.
Voraussetzung für eine optimale Erholung nach Operation (Rekonvaleszenz) und eine möglichst günstige Prognose ist, dass die Diagnose und Indikation (Entscheidung) zur Operation frühzeitig gestellt wird, das Karzinom radikal im Gesunden entfernt wird (sog. R0-Resektion) und der Eingriff ggf. organsparend ausgeführt werden kann.
Nachbehandlung:
Bei R1- und R2- Resektion (mikroskopisch / makroskopisch nicht radikaler Tumorentfernung) erfolgt eine lokale Strahlentherapie, wenn eine Nachresektion nicht möglich ist.
Bei intraoperativem Nachweis eines Befalls der mediastinalen Lymphknoten (Mittelfell, N2-Situation) schließt sich eine Strahlen- bzw. Chemotherapie an.
Der postoperative Verlauf und Rehabilitation
In der Regel ist mit einem Aufenthalt nach Operation von 10-12 Tagen zu rechnen. Nach der Operation steht die frühe Mobilisation, eine konsequente Atemgymnastik (Atem-Coach) und Sekretolyse durch Inhalationen im Vordergrund. Jeder Patient benötigt zur Entlüftung der operierten Thoraxseite eine Sog-Drainage mit Sekretauffangbehälter. Bei unkomplizierten Verlauf kann diese am 1. bzw. 2. postoperativen Tag entfernt werden. Eine rückenmarksnahe Schmerzmittelpumpe (Peridual-Katheter) in den ersten 3-5 postoperativen Tagen plus analgetische Standard-Medikation ermöglicht eine Mobilisation und schmerzfreies Training trotz Thoraxdrainage bereits am 1. postoperativen Tag. Eine eventuelle Nachbehandlung wird mit den beteiligten Ärzten besprochen.
Nach einer Thorakotomie ist normalerweise eine leichte körperliche Arbeit ab der 4.-6. und eine schwere körperliche Arbeit ab der 12. postoperativen Woche vertretbar.
Eine Keilresektion, Segmentresektion oder Lobektomie führt bei präoperativ normaler Lungenfunktion kaum zu einer merkbaren Beeinträchtigung der Atmung. Die Patienten können meistens wieder ihre berufliche Tätigkeit nach Abklingen der Thorakotomiebeschwerden ausüben.
Nach Entfernung einer ganzen Lungenseite (Pneumektomie) oder bei einer ausgeprägten postoperativen Reduktion der Lungenfunktion muss im Einzelfall zur Arbeitsunfähigkeit Stellung genommen werden. Die endgültige Konsolidierung (Ausheilung) der Pneumektomiehöhle mit kompensatorischer Vergrößerung des gegenseitigen Lungenflügels ist erst nach 12-18 Monaten vollzogen. In dieser Zeit geschieht auch die kreislaufmäßige Umstellung auf die neue kardiopulmonale (Herz-Kreislauf-) Situation. In den ersten 6 Monaten ist der Patient nur gering belastbar. Nach dieser Zeit kann körperlich wenig belastende Arbeit verrichtet werden. Eine körperlich schwere Arbeit ist nach der Pneumektomie meistens nicht mehr zumutbar.
Der Sozialdienst unserer Klinik organisiert auf Wunsch den weiteren Rehabilitationsverlauf mit Anschlussheilbehandlung (AHB) bzw. die Bereitstellung einer ambulanten Hilfe zu Hause in den ersten 2 Wochen nach Entlassung. Eine AHB wird in aller Regel für 3 Wochen bewilligt, muss aber spätestens 2 Wochen nach Entlassung begonnen werden.
Mediastinaltumore
Mediastinaltumore (Tumore des Mittelfellraumes)
Die thoraxchirurgische Operation hat heutzutage einen festen Stellenwert im Therapiekonzept mediastinaler Tumorerkrankungen. In Abhängigkeit von der Symptomatik der Patienten, Lokalisation des Tumors im vorderen, mittleren oder hinteren Mediastinum (Mittelfellraum), und Tumorgröße können im Einzelfall zunächst diagnostische Operationen wie die Mittelfellspiegelung (siehe Mediastinoskopie), parasternale Mediastinoskopie oder videoassistierte Brustkorbspiegelung (Thorakoskopie) zur Histologiesicherung (Entnahme einer Gewebeprobe/pathologische Untersuchung) und Therapieplanung indiziert (angezeigt) sein.
Andernfalls wird bei einem primär operablen Befund (durch Operation entfernbarem Tumor) und / oder benigner Dignität (gutartiger Tumorerkrankung) die radikale Tumorexstirpation (Entfernung des Tumors im Gesunden) durchgeführt.

Anatomische Abschnitte
Tumorarten
häufige Indikationen (Entscheidungen) zur Operation - unter Respekt der Lokalisation – sind gutartige Tumoren im:
vorderen Mediastinum: Teratom, Dermoidzyste (Keimzellgeschwülste, Abb.2), Thymom (Thymustumor, Abb.1), retrosternale Strumen (hinter dem Brustbein gelegene Schilddrüsenanteile)
mittleren Mediastinum: bronchogene oder Perikard-Zysten
hinteren Mediastinum: Neurinome (Nervengeschwülste, Abb. 3)
Bei der "Myasthenia gravis" (Muskelermüdungserkrankung) ist die Entfernung des Thymus, in der Regel thorakoskopisch, ein fester Bestandteil des Therapiekonzeptes.
Tabellarische Übersicht der Tumorarten nach der anatomischen Lage:Vorderes und oberes Kompartiment: Mittleres (axiales) Kompartiment: Hinteres Kompartiment:
- Thymome
- Schilddrüsen-....neoplasien
- Lymphome
- Lymphknoten- ....metastasen
- Keimzelltumoren
- Paragangliome
- Pleurapericard-....cysten
Lymphome
Lymphknoten-metastasen
Paragangliome
Bronchogene Zysten
Oesophagus-
divertikel
- Neurogene Tumoren:
Schwanome
Neurofibrome
Ganglioneurome
Neuroblastome
- Paragangliome
- Gastroenterale ...Zysten
Abb.: Perikardzyste
Abb. 1: Operationsfoto eines Thymom im vorderen Mediastinum
Selten werden auch Karzinome und Sarkome (maligne Tumoren, Krebsgeschwülste) im Mediastinum operiert.
Abb. 2 Magnetresonanz-Tomographie-Bilder (MRT): Neurinom im hinteren Mediastinum
, Abb. 3 Magnetresonanz-Tomographie-Bilder (MRT): Dermoidzyste im vorderen Mediastinum
Mediastinaltumore werden in der Regel über einen Sternotomie-Zugang (Längsspaltung des Brustbeines) oder via lateraler Thorakotomie (seitliche Brustkorberöffnung, im Rippenzwischenraum), in Einzelfällen auch über eine videoassistierte Thorakoskopie (Thymomektomie) operiert und entfernt. Bei Infiltration (Einwachsen) der Tumore in die Nachbarorgane oder –Gewebe können diese Strukturen (z.B. Lunge, Perikard, Pleura, Gefäße) anteilig mit entfernt werden, um eine Resektion (Entfernung) des Tumors im Gesunden zu erreichen. Defekte können hierbei durch Kunststoff-material (z.B. Gortex) ersetzt und damit die Organstruktur wiederhergestellt werden (z.B. Kunststoff-Implantation und Wiederherstellung großer Gefäße).
Im Einzelfall kann es bei malignen oder zweifelhaften Tumoren erforderlich sein, ergänzend zur Operation, noch eine Chemotherapie und / oder Strahlentherapie in das Therapiekonzept einzubinden. Ziel ist es dabei, für den individuellen Patienten eine möglichst definitive und langzeitige Heilung zu erreichen.
Lunge und MediastinumBronchialkarzinom mediastinale Tumore Pleuramesotheliom
[link:9]
[/link:9]
[/link:9]
Pleuramesotheliom (Karzinom des Rippenfells)
Die Inzidenz des malignen Pleuramesotheliom beträgt ca. 1 : 1 Mio. Einwohner pro Jahr. Der Tumor geht vom Epithel der viszeralen und parietalen Pleura aus. In ca. 50-90% ist eine Asbestexposition in der weit zurückreichenden beruflichen Vorgeschichte nachweisbar. Das asbestinduzierte Pleuramesotheliom ist eine entschädigungs-pflichtige Berufskrankheit. Betroffen sind hauptsächlich Männer im 6.Lebensjahrzehnt, die in sog. Staubberufen tätig waren. Trotz zunehmender Elimination der Asbestfasern als Baustoff seit den 70 er Jahren wird wegen der langen Latenzzeit mit einem weiteren Anstieg der Erkrankungsfälle bis zum Jahre 2017 gerechnet.
Die Erkrankung folgt mit einer Latenz (Abstand zwischen Exposition und Krankheit) von ca. 25-40 Jahren. Leitsymptome sind der Thoraxschmerz und der Pleuraerguß.
Insgesamt ist die Prognose bei auftretenden Beschwerden schlecht (mittlere Überlebenszeit: 7 - 14 Monate). Die therapeutischen Möglichkeiten sind äußerst begrenzt. Es überwiegen palliative (lindernde) Maßnahmen zur Verminderung der Ergußproduktion und der Schmerzlinderung. Eine Operation im asymptomatischen Frühstadium ist lebensverlängernd
Abb.
Thorakoskopisch Pleuramesotheliom
prognostisch ungünstige Faktoren sind:
nicht-epithelialer Zelltyp
schlechter Allgemeinzustand
männliche Geschlecht
hohe Leukozytenzahl (weißen Blutplättchen)
niedriger Hämoglobinwert
Abb:
konventionelles Thorax-Röntgen Bild bei Pleuramesotheliom der linken Lunge
Bildgebende Verfahren
Häufigste Manifestation in den bildgebenden Verfahren (Röntgenthorax, Ultraschall) ist der Pleuraerguß. Die durch Punktion gewonnene Flüssigkeit zeigt in der zytologischen Untersuchung lediglich in ca. 50% positive Ergebnisse, vor allem beim epithelialen Subtyp. Das CT spielt für die Beurteilung der Tumorausdehnung die wichtigste Rolle,
Abb:
Thorax-CT bei Pleuramesotheliom der linken Lunge
Die histologische Diagnosesicherung erfolgt durch die Thorakoskopie mit gezielter Probeentnahme (Abb.). Die diagnostische Ausbeute liegt bei ca. 95%. Außerdem erlaubt sie gleichzeitig ein Staging (Ausdehnung des Befalls), da der Befall von Zwerchfell, Lunge, Perikard usw. makroskopisch und nach Probeentnahme auch mikroskopisch nachgewiesen bzw. ausgeschlossen werden kann. Trotz der Neigung des Mesothelioms, sich entlang von Punktionskanälen und Drainageeintrittsstellen auszubreiten (sog. Impfmetastasen), sollte die invasive Diagnostik zur histologischen Sicherung immer durchgeführt werden.
Abb.: Thorakoskopische Gewebeprobenentnahme bei Pleuramesotheliom
Therapie:
Die therapeutischen Möglichkeiten sind äußerst begrenzt. Nur in den sehr frühen Stadien (T1+T2, selektioniert T3) ist eine Indikation zu radikalchirurgischen Maßnahmen als extrapleurale Pleuro-Pneumonektomie einschließlich Resektion von Perikard und Diaphragma (Pleuropneumo-perikardiodiaphragmatektomie (P3D) mit adjuvanter Chemotherapie gegeben. Das Mesotheliom ist gegen eine Bestrahlung weitgehend resistent. Die Radiotherapie dient hauptsächlich zur Schmerzreduktion oder zur Reduzierung extrathorakaler Tumormassen im Bereich von Stichkanälen. Als wesentliche palliative Maßnahme wird zur Verminderung der Ergußproduktion eine Pleurodese mit sklerosierenden Substanzen (z.B. Talkum) durchgeführt.
Operationsverfahren beim Pleuramesotheliom sind
Diagnostisch:
Thorakoskopie (VATS)
Palliativ:
Talkumpleurodese
Palliative parietale Pleurektomie
Potentiell kurativ:
Extrapleurale (erweiterte) Pleuropneumektomie (P3D)
Sollte sich intraoperativ der Tumor als nicht resektabel (komplett entfernbar) erweisen, kann innerhalb derselben Operation die parietale Pleurektomie zur Vermeidung rezidivierender Pleuraergüsse palliativ vorgenommen werden. Bei rezidivierenden Ergüssen sollte eine thorakoskopische Talkumpleurodese erfolgen. Bei allen anderen Konstellationen ist zurzeit ein individuelles palliatives Vorgehen zu empfehlen.
Prognose:
Die Prognose des Pleuramesotheliom ist außerhalb kurativer chirurgischer (multimodaler) Ansätze äußerst schlecht. Die mittlere Überlebenszeit nach Symptombeginn beträgt 12-18 Monate. Sarkomatoide Subtypen haben eine schlechtere Prognose als epitheliale Subtypen (in Einzelfällen Überlebenszeiten von 8-9 Jahren).
Nur durch Früherkennung und den frühen Einsatz kurativer Resektionsbehandlungen die Prognose verbessern. Für die Prophylaxe ist die Vermeidung der Asbestexposition entscheidend.
Stand Juni 2008
Verfasser:
Dr. Carsten Vogel
ehem. Klinik für Allgemein-, Viszeral-, Thorax-, Transplantations- und Kinderchirurgie
Uni-Klinikum Gießen/Marburg, Standort Gießen
Rudolf-Buchheim-Str. 7 35385 Gießen
Telefon: 0641 99 44710
Telefax: 0641 99 44709
eMail: carsten.vogel@chiru.med.uni-giessen.de
eSite: Chirurgie Univ. Gießen
überarbeitet/ aktualisiert 03-2016
Lymphödem bei Brustkrebs
1. Einleitung:
Das sekundäre Lymphödem bei Brustkrebs
ist ein chronisches, teilweise zur Progression neigendes
Krankheitsbild. Es entsteht durch eine Schädigung der Armlymphgefäße im
Armwurzelbereich mit dadurch bedingter Störung im Lymphabfluss infolge
operativer Entfernung oder radiogener Fibrosierung von axillären oder
klavikulären Lymphknoten. Entsteht eine solche Lymphstauung durch eine
metastatische Blockade in den Lymphknoten wird das Ödem als Malignes
Lymphödem bezeichnet. Lymphödeme bei Mamma-Ca. können Arm-, Hand-,
zugehörige Brustwand und Brustdrüse betreffen. Das Lymphödem
manifestiert sich meist sofort nach der Operation, es kann jedoch auch
infolge Narbenschrumpfung erst Monate bis Jahre später auftreten, was
besonders nach einer axillären Radiatio beobachtet wird.
Infolge der Lymphabfluss-Sörung
sammelt sich eiweißreiche Flüssigkeit im subkutanen interstitiellen
Bindegewebe, wodurch die für Lymphödeme typischen Proteinfibrosen im
Laufe von Monaten bis Jahren entstehen.
Durch verbesserte Operationsverfahren
und schonendere Bestrahlung ist die Morbidität des Lymphödems in den
letzten Jahrzehnten deutlich gesunken und wird zz. auf etwa 5 %
geschätzt. Ein Mamma-Ödem nach Bestrahlung der Restbrust ist dagegen
häufiger. Dabei handelt es sich jedoch nicht um ein echtes Lymphödem,
sondern um ein durch radiogene Schädigung der Blutkapillaren bedingtes
„entzündliches Ödem“, welches sich in der Regel innerhalb von höchstens 2
Jahren komplett zurückbildet. Dieses Mamma-Ödem muss von einer
Radiofibrose der Brust differenziert werden, welche persistierend ist.
Weiterhin darf ein Lymphödem nicht mit der häufigen postoperativen
traumatischen Ödematisierung im Oberarm-, Schulter- und angrenzendem
Thoraxwandbereich verwechselt werden, welche sich normalerweise nach
einigen Wochen bis Monaten spontan zurückbildet. Bei einer
Wächter-Lymphknoten-Op. ohne Entfernung von axillären Lymphknoten ist
nicht mit einem Lymphödem zu rechnen.
2. Lymphödemstadien:
Stadium 1:
reversibles Lymphödem, spontan oder infolge Therapie, keine Gewebeveränderungen der Haut.
Stadium 2:
manifestes (irreversibles) Lymphödem
mit leichtgradigen Komplikationen: subkutane Proteinfibrose evtl.
Hyperkeratose oder Papillomatose. Stad. 2 entspricht dem typischen
Lymphödem.
Stadium 3:
manifestes (irreversibles) Lymphödem
mit schwerwiegenden Komplikationen: massive subkutane Proteinfibrose,
Pachydermie, Hyperkeratose, Papillomatose, Nagelbettveränderungen,
Lymphzysten, Lymphfisteln, Ekzeme, Ulzerationen, häufige Erysipele,
Angiosarkom.
3. Lymphödemschweregrade:
- Grad I
- = geringes Ödem: Ödemvolumen bis 25 % *
- Grad II
- = mäßiges Ödem: Ödemvolumen bis 50 % *
- Grad III
- = starkes Ödem: Ödemvolumen bis 100 % *
- Grad IV
- = massives Ödem: Ödemvolumen bis 200 % *
- Grad V
- = gigantisches Ödem: Ödemvolumen über 200 % *
*gegenüber der gesunden Seite
Bestimmung des Ödemgrades bei einseitigem Lymphödem mit dem
Ödemgradmesser. Bei beidseitigen Armlymphödemen muss der Ödemgrad im
Verhältnis zu einer fiktiven Normal-Extremität geschätzt werden.
4. Lymphödemsymptome:
Schwellung, Schweregefühl, Bewegungsbehinderung, Leistungsverminderung, Spannungsschmerzen, psychische Belastung.
5. Lymphödemdiagnostik:
Es ist eine klinische Diagnose, welche
durch Anamnese und körperliche Untersuchung gestellt wird. Die
Beurteilung und Dokumentation geschieht am einfachsten über
Umfangmessungen, welche präoperativ, am Ende der stationären Behandlung
und jeweils bei den Tumornachsorgeuntersuchungen an beiden Armen
durchgeführt werden sollten. Als Messpunkte empfehlen sich der Umfang
der Mittelhand (hier ist der Ödemgradmesser nicht indiziert), am
Handgelenk, an der umfangstärksten Stelle am proximalen Unterarm und in
Oberarmmitte. Weiteres diagnostisches Kriterium ist die verdickte
Hautfalte infolge subkutaner Eiweißfibrose und die Dellbarkeit. Bei
plötzlichem Auftreten einer Armschwellung ist auch an eine venöse
Thrombose zu denken, besonders wenn die Schwellung mit einer Zyanose und
verstärkter Venenzeichnung einhergeht. Dann ist eine entsprechende
phlebologische Diagnostik erforderlich.
Bei V.a. ein malignes Lymphödem
(Ödemschwerpunkt am Oberarm, Übergreifen des Ödems auf den angrenzenden
Rumpfquadranten, progrediente Ödemverschlechterung, zunehmende
Armplexusschädigung, Venektasien an der Extremitätenwurzel, venöser
Umgehungskreislauf der Schulter, Überwärmung des Ödems, Lymphangiosis
carcinomatosa cutis) ist eine Rezidivdiagnostik erforderlich.
6. Lymphödemkomplikationen:
- Regelmäßig auftretende Komplikation:
- Proteinfibrose
- Häufig auftretende Komplikationen:
- Erysipel, Weichteilrheumatische Beschwerden
- Selten auftretende Komplikationen:
- Ekzem, Lymphzyste, Lymphfistel, Papillomatose der Haut
- Extrem seltene Komplikationen:
- Lymphogenes Ulcus, Angiosarkom.
7. Lymphödemtherapie:
Therapieziele:
- Reduzierung des Ödems und seiner Beschwerden,
- Reduzierung von Ödemkomplikationen,
- Erhaltung oder Wiederherstellung der Funktion, Leistungs- und
Arbeitsfähigkeit mit dem Arm.
Therapiemöglichkeiten:
- 1. Physikalische Ödemtherapie = Komplexe physikalische Entstauung (KPE)
- 2. Operationen
zu 1. Physikalische Ödemtherapie = Komplexe physikalische Entstauung (KPE):
· Kombination von Manueller Lymphdrainage (MLD) und Kompression.
· Zusammenarbeit Arzt–Lymphtherapeut–Sanitätshaus wichtig.
· Physikalische Ödemtherapie kann
ambulant bei Lymphtherapeuten (Masseure, medizin. Bademeister,
Krankengymnasten) oder stationär in lymphologischen Fachkliniken
(Ödemkliniken) durchgeführt werden. Ambulant vorzugsweise bei
leichtgradigen Ödemen, bei denen keine wesentliche Ödemabnahme zu
erwarten ist oder im Anschluss an eine stationäre lymphologische
Behandlung als Erhaltungstherapie. Adressen von Lymphtherapeuten unter www.Lymphtherapeutenliste.de
· Ambulante Lymphdrainage ist möglich mit 30 Min., 45 Min. oder 60 Min.
- - 30 Min. bei einseitig leichtgradigem Lymphödem,
- - 45 Min. bei bds. leicht- oder einseitig schwergradigem Lymphödem,
- - 60 Min. bei bds. schwergradigen Lymphödemen
MLD soll grundsätzlich bei Ödemen nur
in Kombination mit einer Kompression verordnet werden, meist als
Strumpf. Wird ein Strumpf von einem Patienten abgelehnt, sollte MLD
nicht verordnet werden. MLD ohne Tragen eines Kompressionsstrumpfes ist
Geldverschwendung.
Voraussetzung für die Verordnung von
MLD ist eine Umfangszunahme von mindestens 1 cm mit Ödembeschwerden. Bei
einer Umfangszunahme von über 2 cm sollte auch ohne Beschwerden MLD
verordnet werden.
Richtlininen für die ambulante MLD-Frequenz:
- Geringes Lymphödem:
- MLD 0 – 1x/Woche 30 Min., Strumpf überwiegend tragen.
- Mäßiges Lymphödem:
- MLD 1 - 2x/Woche 45 Min., dauerndes Strumpftragen.
- Starkes Lymphödem:
- MLD 2 - 3x /Woche 45 Min., dauerndes Strumpftragen.
- Massives Lymphödem:
- MLD 3 - 4x/Woche 45 Min., dauerndes Strumpftragen.
- Gigantisches Lymphödem:
- MLD 3 - 5x/Woche 45 Min., dauerndes Strumpftragen.
Stationäre lymphologische Behandlung
ist indiziert bei schwergradigen Lymphödemen zur Ödemreduktion und bei
leichtgradigen Lymphödemen, die auf ambulante Behandlung nicht
ausreichend ansprechen. Stationäre Behandlungen werden in
lymphologischen Fachkliniken oder Ödemkliniken als Rehamaßnahme
durchgeführt und müssen bei der Krankenkasse bzw. beim
Rentenversicherungsträger beantragt werden. Adressen von Ödemkliniken
unter www.dglymph.de àLymphkliniken.
Kompressionstherapie:
- möglich sind Bandagen,
Bestrumpfungen und apparative intermittierende Kompression (AiK).
Bandagen werden vorzugsweise in Ödemkliniken in der Ödemreduktionsphase
eingesetzt.
- Kompressionsstrümpfe werden ambulant benutzt und vor Entlassung aus der Ödemklinik angemessen und ausgehändigt.
- AiK ist sowohl stationär als
auch ambulant möglich. Bei der ambulanten AiK sollte sie als
Heimbehandlung erfolgen. Beim Armlymphödem sind 3-Kammer-Armmanschetten
ausreichend (z.B. Vaso-Flow der Fa. Bösl).
- Bei schwerergradigen
Lymphödemen oder bei Komplikationen, besonders rezidivierenden
Erysipelen, sind erneute stationäre lymphologische Behandlungen auch zur
Erhaltung der Arbeitsfähigkeit erforderlich.
Bestrumpfungen der unterschiedlichen Armlymphödemvarianten
Ödemlokalisation
Bestrumpfung
Finger-Hand-Ödem
Handschuhe mit kurzen oder langen Fingern
Handrückenödem (ohne Fingerödem)
Handschuh ohne Finger
Unterarm-Hand-Ödem
Langer Handschuh bis Ellenbeuge mit oder ohne Finger
Armödem
Armstrumpf
Armbestrumpfungen normalerweise in
Kompressionsklasse 2. Bei Armstrümpfen wird bei Bedarf am oberen
Strumpfrand ein Silikonband gegen das Herunterrutschen eingenäht.
Schulterkappen und Strumpfhalterungen sind normalerweise nicht
erforderlich.
Ergänzende Therapiemaßnahmen:
- Anlernen in der Lymphdrainage-Eigentherapie durch den Lymphtherapeuten,
- Entstauungsübungen für Arme (Anlage 1),
- Armhochlagerung,
- Patientenschulung durch Verhaltensregeln Arm (Anlage 2),
- lymphologische Betreuung.
zu 2. Operationen bei Lymphödemen:
Theoretisch sind 2 operative Verfahren denkbar:
- Bei der Lymphgefäßtransplantation wird ein Lymphkollektor vom Oberschenkel als Umgehung der zerstörten Achsellymphbahnen subkutan mikrochirurgisch implantiert.
- Bei der lymphovenösen Anastomose werden gestaute Lymphgefäße an Venen anostomosiert.
In beiden Fällen soll der Lymphabfluss
verbessert werden. Die Erfahrungen mit diesen Operationen sind nicht
groß und nicht gut, so dass sie nur in Fällen von Versagen der
physikalischen Ödemtherapie in Betracht gezogen werden sollten.
8. Kontraindikation für MLD:
- Akute Axillarvenen- oder Subklaviavenenthrombose,
- akute Armentzündung, besonders Erysipel,
- akutes Ekzem am Ödemarm,
- V.a. alleiniges lokales oder lokoregionales Tumorrezidiv,
- dekompensierte Herzinsuffizienz,
- Halsbehandlung bei Hyperthyreose oder Struma nodosa.
9. Lymphödem-Prophylaxe:
- Die Aufklärung des Pat. über das Lymphödemrisiko muss grundsätzlich in der operierenden und bestrahlenden Klinik erfolgen.
- Aushändigung der Verhaltensregeln bei Armlymhödem (www.dglymph.de àmedizin. Infos àVerhaltensregeln Arm), Anlage 2.
- Prophylaktische MLD oder prophylaktisches Tragen eines Armstrumpfes ohne Lymphödem ist nicht sinnvoll und ökonomisch.
- Allenfalls kann das postoperative traumatische Ödem im Armwurzelbereich vorüberghend mit 1-2x wöchentlicher Lymphdrainage von 30-45 Min. bis zu 3 Monate behandelt werden.
10. Sozialmedizinische Beratung:
Die Arbeitsfähigkeit kann bei einem
Armlymphödem entsprechend der Händigkeit erheblich reduziert sein. Mit
einem Armlymphödem sollen keine mittelschweren körperlichen Arbeiten
sowie keine monotonen leichten Arbeiten durchgeführt werden, evtl.
Teilrente erwägen.
Schwerbehinderung für ein leichtgradiges Armlymphödem 0-10 %, wenn am Führungsarm
20 %, für ein schwergradiges
Armlymphödem mit geringer Leistungseinschränkung 30 %, wenn am
Führungsarm 40 %. Bei bds. leichtgradigen Armlymphödemen 20 %. Bei bds.
schwergradigen Armlymphödemen 40-60 %.
11. Psychologische Betreung:
In Fällen von gestörter Krankheitsverarbeitung ist eine psychologische Betreuung erforderlich.
12. Hinweise auf Selbsthilfegruppen:
Vermittlung an Selbsthilfegruppen ist
oftmals erforderlich, besonders „Frauenselbsthilfe nach Krebs“. Adressen
über den Bundesverband unter www.Frauenselbsthilfe.de oder Tel. 0 60 21-24 43 4.
Beratung hinsichtlich Brustprothesen,
Lymphentlastungs-BH und entsprechender Kleidung durch Sanitätshäuser und
Selbsthilfegruppen.
Über Selbsthilfegruppen auch Vermittlung von Gruppenentstauungsgymnastik und Sport nach Krebs.
- Autor: Dr. Ulrich Herpertz
- Internist, Lymphologe
- Generalsekretär der Deutschen Gesellschaft für Lymphologie
- www.lymphforum.de
- E-Mail: dr.ulrich@herpertz.net
Verlinkung
zum
Krebsinformationsdienst Heidelberg

Bauchspeicheldrüsenkrebs | Brustkrebs | Darmkrebs | Gebärmutterhalskrebs
Gebärmutterkörperkrebs | Harnblasenkrebs | Leukämien
Lungenkrebs | Lymphome | Magenkrebs
Prostatakrebs